
Welcome to the “Coffee break with TRANSFAC”
A series of online sessions hosted by Dr. Alexander Kel, CEO geneXplain GmbHThe next Coffee break with TRANSFAC will be held on December 17th 6 PM CET
Fill in this form to receive the event joining link
From Zebrafish to Precision Medicine: Discovering Master Transcription Factors with TRANSFAC®
In this webinar Prof. Dr. Alexander Kel, the CEO and CSO of geneXplain GmbH, will demonstrate how TRANSFAC® and the tools included in the geneXplain platform enable the identification of key transcription factors in model organisms like zebrafish, bridging the gap between basic research and medical applications for drug discovery.
The following main questions will be in the focus of this seminar:
About the Coffee breaks with TRANSFAC
This initiative of Q&A sessions with a leading bioinformatics expert Dr. Alexander Kel is intended to support all researchers out there that are interested in the area of applied bioinformatics.
Come to our sessions as a simple listener, or become an active participant of this recurrent event and ask Dr. Kel your own questions in order to emphasise the direction of the live discussion.
You can send your questions via the form below or by email: [email protected] with a subject “Question to Dr. Kel”.
Questions can also be asked live during the online event.
Ask your question by filling in this form:
[fluentform id=”17″]
What to ask about?
Any question that you need assistance with while performing your bioinformatics analysis, e.g.
- Promoter analysis? Pathway analysis?
- Can AI analyze NGS data?
- How to combine DNA methylation and metabolome data?
- What to start from and how to interpret the obtained results?
and much more….
Check out the video records of the previous “Coffee break with TRANSFAC” sessions
- 26 November 2024, CRISPR Synthetic Lethality screens and TRANSFAC Pathways
- 12 November 2024, “Intelligent design” of promoters: Practical Guide
- 29 October 2024, DNA methylation signatures of diseases
- 15 October 2024, TRANSFAC Q&A Session
- 8 October 2024, “Intelligent design” of promoters
- 20 August 2024, ATAC-seq, CUT&RUN, and Enhancers
- 9 July 2024, Release 2024.1 new features overview
- 18 June 2024, Don’t do pathway analysis without TRANSFAC
- 28 May 2024, Download TRANSFAC and do whatever you like
- 21 May 2024, How to find tissue-specific transcription factor target genes?
- 14 May 2024, ATAC-seq, CUT&RUN and Enhancers
- 30 April 2024, geneXplain platform API
- 22 April 2024, Promoter analysis of plants
- 16 April 2024, Your first TRANSFAC analysis
- 8 April 2024, Exploring the FREE geneXplain platform account
- 2 April 2024, Your income and your genes
- 19 March 2024, MATCH Suite software demo
- 12 March 2024, Promoter analysis of model organisms (part 2)
- 4 March 2024, Promoter analysis of model organisms (part 1)
- 6 February 2024, How to find tissue-specific TF target genes
- 23 January 2024, Your first TRANSFAC analysis
- 12 December 2023, How to find TRANSFAC motifs in ATAC-seq peaks? (part 2)
- 30 November 2023, How to find TRANSFAC motifs in ATAC-seq peaks? (part 1)
- 7 November 2023, TRANSFAC pathways. How to find them?
- 10 October 2023, What is the way from TFs to pathways?
- 19 September 2023, How to build gene regulatory networks using TRANSFAC, Jupyter Notebook and API
- 8 June 2023, Master-regulators of Glioblastoma pathways – special session dedicated to the World Brain Tumor Day
- 25 May 2023, Promoter analysis of model organisms
- 25 April 2023, How to apply TRANSFAC for analysis of cancer mutations
- 4 April 2023, How to find motifs created or destroyed by SNPs
- 23 March 2023, De novo motifs: how to find and use them
- 2 March 2023, How to compute transcription factor binding affinity
- 7 February 2023, How to find a list of tissue-specific TF target genes
- 17 January 2023, Promoter analysis? Pathway analysis? What to start from? How to interpret the results?
CRISPR Synthetic Lethality screens and TRANSFAC Pathways
26 November 2024, the thirty fourth Coffee break with TRANSFAC” session
Click here to view the event details
Your questions addressed within this session:
01:22 Synthetic lethality – what is it?
03:17 CRISPR library used for synthetic lethality screens
08:24 PD-L1 knockout integrated analysis of CRISPR screening (GSE268533) and CUT&Tag data (GSE268534)
12:54 General Mechanism of immune therapy action (immune checkpoint inhibitors of PD-L1)
16:47 Basic principles of CUT&Tag analysis. Identification of places where TFs are binding on the DNA.
18:33 Finding co-factors for PD-L1 with the use TRANSFAC PWMs and combinatorial analysis of sites. Combinations of transcription factors bringing synergistic effect.
22:06 Overview of the algorithm for integrative analysis of CUT&Tag and CRISPR synthetic lethality screens data. Identification of master regulators.
26:31 Analyzing data from GSE268533 and GSE268534 using the geneXplain platform and Genome Enhancer. Positive and negative scores of genes (in case of knockout, the gene either helps the cell to survive or pushes it to death)
36:18 CUT&Tag data is showing where PD-L1 is located in the genome. Finding genes located near the peaks of PD-L1. Converting genes with high negative scores from synthetic lethality screens to proteins for further usage in Genome Enhancer as proteomics data
42:27 Launching Genome Enhancer analysis on the obtained epigenomics data (CUT&Tag) + proteomics data (generated from synthetic lethality screens data) + list of genes with maximum number of peaks in their vicinity (used as “transcriptomics” data in the Genome Enhancer pipeline) in the context of head and neck neoplasms and squamous cell carcinoma
47:09 Looking into the results produced by the Genome Enhancer pipeline. Combinations of transcription factor binding sites found in the peaks. Constructed network connecting all analyzed data types together. Identified prospective drug targets and associated drugs.
01:03:01 How to find out the functionality of a given gene set on an example of the set of genes encoding master regulators that were found. GSEA. Using your own ontology for functional classification (e.g. uploading data from MSigDB; upload of your own ontology in the format of gene lists; calculation of the p-values and FDRs of overlaps between the input gene list and the gene lists from the uploaded ontology folder)
“Intelligent design” of promoters: Practical Guide
12 November 2024, the thirty third “Coffee break with TRANSFAC” session
Click here to view the event details

Your questions addressed within this session:
00:52 Introduction
02:09 Gene regulatory code and basic principles of gene regulation
07:46 Prediction of transcription factor binding sites
08:31 Areas of artificial promoters application
10:02 Artificial promoters application in tissue and organ engineering
13:50 Optogenerapy project: Optegenetic Protein Therapy for Multiple Sclerosis. Designing an implant (device) with genetically engineered cells, in which signaling that turns on specific genes is switched on by the red light
15:46 Artificial promoters application in synthetic biology
17:12 Heart construction using iPSCs
19:28 Artificial promoters application in cancer therapy
25:41 Application of AI in generating artificial promoters. What is the “alphabet” and what are the “words”? “Emotions” that can be recognized in promoters.
38:57 Understanding the structure of enhancers and promoters that are active upon RUNX1 activation on the basis of multi-omics data analysis (GSE120216 RNA-seq dataset of RUNX1 knockout in tumor cells after 6 hours and GSE129314 ChIP-seq dataset of RUNX1 knockout in tumor cells at various time points)
44:31 Login to the geneXplain platform / Genome Enhancer. Upload and processing of data from GSE120216 and GSE129314 in the system
48:51 Uploading RNA-seq data (table with read counts) to Genome Enhancer
50:17 Uploading ChIP-seq data (table with peaks) to Genome Enhancer
53:25 Preparing ChIP-seq data for further analysis: converting the table with peak coordinates to a track
57:21 Visualization of the obtained track with peaks in the genome browser
59:43 Launching the analysis in Genome Enhancer: specifying the analysis conditions
01:04:43 Viewing the constructed enhancer model in Genome Enhancer analysis results
01:07:09 Viewing the enhancer model of EYA2 gene (most downregulated gene upon inhibition of RUNX1). Creating artificial construct containing both: enhancer and promoter of EYA2
01:09:02 Genome browser overview of EYA2 gene. Improving the promoter of EYA2: increasing the scores of TFBS of RUNX1 and GR (duplicating the binding sites of AML1 and GR; deleting gaps between sites)
01:11:40 Analyzing the constructed promoters using the “Site search on track” tool
01:14:12 Applying the model of promoter and enhancer created by Genome Enhancer to the track of found sites
DNA methylation signatures of diseases
29 October 2024, the thirty second “Coffee break with TRANSFAC” session
Click here to view the event details
Your questions addressed within this session:
01:33 Epigenomics: “rewriting the book” of the genomics
04:02 Chemical basis of DNA methylation
04:59 Why DNA methylation is important
06:09 Effect of DNA methylation on gene regulation
11:00 CpG islands and their frequency among the genome
12:17 Gene regulation machinery and DNA methylation effects on it
17:48 Do TFs bind to the same motifs regardless of methylation status of respective oligonucleotides? (data from this paper)
24:17 Outline: why it is important to analyze individual CpG sites and not whole (de)methylated regions
25:19 Diseases affected by DNA methylation changes
27:59 How DNA methylation changes can be technically detected. Bisulfate sequencing, methylation arrays
31:32 How differentially methylated regions can be analyzed. DNAm influence on gene regulation. Estimation of binding sites frequency and enrichment around CpG positions
40:11 KABUKI syndrome – a rare genetic disease. Analyzing data from GSE97362 in Genome Enhancer and the geneXplain platform
01:02:08 Is DNA methylation pattern good to be studied by blood samples?
01:05:17 How to interpret DNA 5mC methylation change at the single nucleotide level? Does it have significant impact on the gene and disease when it’s super rare and in a diseased patient?
01:08:53 Online tools for in silico DNA methylation analysis?
01:10:08 Is DNA methylation mapping of each individual unique like a finger print or are there some common methylation regions across the genome of each individual?
01:12:00 Interesting to observe that methylation might favor the transcription factor Hox and repress Oct4. Do we know if there are any or no methyl groups added, like in histone we have me1, me2, me3?
01:14:55 A vast number of lifetime events and extraneous factors in every individual’s lifespan impact methylation in the human genome, many may have lasting effects on overall health and some may be the underpinnings for major diseases. When studying epigenomics in the brains of patients who died of suicide or others who had schizophrenia or bipolar, how do we account for a myriad of undocumented vagaries that may have subtle causal role in the overall disease outcome? Some of the true causal perpetrators are never traceable under the heavy burden of medication effects, toxicology, and other comorbid conditions, etc. I would like to know your take on this.
01:19:25 Which changes occur in the methylation of some genes during the meditation, or on histones?
01:21:04 How can we solve the gene-specific hypomethylation to treat cancer?
01:24:01 Recommended workflow to analyze DNA methylation data?
01:30:53 How to integrate other omics data (RNA-seq, proteomics) together with Methylation data if available?
TRANSFAC Q&A Session
15 October 2024, the thirty first “Coffee break with TRANSFAC” session
Click here to view the event details

Your questions addressed within this session:
00:15 Brief introduction to the TRANSFAC database
04:56 Which transcription factors are regulating my genes?
06:50 Which transcription factors are regulating my genes – reply based on the TRANSFAC knowledge base
12:13 Which transcription factors are regulating my genes – prediction made using the MATCH Suite tool. Tissue and functional specificity of TFs.
27:57 Are there modified forms of transcription factors in TRANSFAC?
30:50 Are there pathways upstream of TFs included in TRANSFAC, e.g. in Full package?
33:09 Pathway visualization in PathFinder
34:13 Can you search for specific miRNAs in TRANSFAC?
38:36 Is the TRANSFAC module included in the geneXplain platform?
41:23 What kind of information is covered in the “mutations” section of the result page when a specific TF is searched? (Is it just specific to a disease condition/somatic mutation/germline mutation?)
44:53 Can you study the transcriptional regulation of any gene e.g. NRF2 and its binding partners in any condition?
“Intelligent design” of promoters
8 October 2024, the thirtieth “Coffee break with TRANSFAC” session
Click here to view the event details
Your questions addressed within this session:
01:18 Regulatory code: what do we know about it and why is it needed
05:21 Areas of artificial promoters application
11:30 Dynamics of synthetic biology and regenerative medicine technologies development
15:05 Synthetic biology and regenerative medicine tools. Approaches to gene regulation using artificial promoters.
19:59 Promoter design in tissue and organ engineering
22:13 Promoter design in cancer therapy
28:37 Using AI to generate promoters and enhancers
38:33 Synthetic promoters and their construction using AI and TRANSFAC
45:39 Genome Enhancer application for building a model that discriminates the promoters of upregulated genes in glioblastoma cell lines under hypoxia condition from the promoters of the non-changed genes (GSE232725)
50:01 Designing new promoters: example of AML1 (RUNX1) data analysis from integrated ChIP-seq and RNA-seq dataset (GSE129314 and GSE120216). Design of EYA2 gene promoter based on the input FASTA file.
1:00:43 Conclusion: the complexity of the gene regulation code
1:06:01 We can now create artificial blood vessels, skin, and even noses. But what challenges remain in growing more complex organs like the liver or heart? How much more difficult is it, and what steps must we take to achieve this breakthrough?
1:09:50 While designing artificial promoters to impact the gene expression, are SNPs in the core promoter or upstream regulatory regions considered as a “potential target” and given priority or only the modification of a given sequence in these regions is taken into account to create such promoters?
ATAC-seq, CUT&RUN and Enhancers
20 August 2024, the twenty ninth “Coffee break with TRANSFAC” session
Click here to view the event details

Your questions addressed within this session:
00:49 Gene regulation principles. Epigenetic regulation of gene expression.
04:17 Experimental methods for TF binding analysis. ChIP-seq, ATAC-seq, CUT&RUN.
10:45 Processing of ChIP-seq, ATAC-seq, CUT&RUN data. Peak calling algorithms.
15:22 Search for enriched TFBS in peaks.
17:36 geneXplain platform for analysis of ChIP-seq, ATAC-seq, CUT&RUN data
19:01 Example analysis of ChIP-seq data generated from HeLa cells with antibody to ETF1 (fastq data analysis from GSM558469)
21:49 Alignment of fastq file: mapping to genome using Bowtie2
23:50 BAM file visualization on genome browser
25:16 Peak calling with MACS. Finding clusters of peaks
30:09 Search for enriched TFBS in clusters of peaks
40:00 Example analysis of ChIP-seq, CUT&RUN and transcriptomics data generated for B-cell lymphoma before and after FOXO1 inhibition (data from GSE254405)
44:31 Modules of transcription factors working together: CMA for search of binding site compositions
45:49 How much time will an analysis take from data loading, e.g. 2GB, to receiving a result?
46:38 Applying Genome Enhancer for analysis of data from example GSE254405 (integrated CUT&RUN and transcriptomics data analysis)
51:20 Algorithm underlying the integrated analysis of transcriptomics and epigenomics data in Genome Enhancer
53:43 Analysis launch interface in Genome Enhancer
56:56 Genome Enhancer analysis report overview
01:02:51 Can we use this analysis in plant science research to develop climate-resilient crops
01:06:10 For peak calling of CUT&RUN histone reads, is SEACR better than MACS in your experience?
01:09:00 Can we use this analysis for microbe genomes?
1:12:12 What are the limitations of CUT & RUN Seq Analysis
Release 2024.1 new features overview
9 July 2024, the twenty eighth “Coffee break with TRANSFAC” session
Click here to view the event details

Your questions addressed within this session:
00:37 TRANSFAC release 2024.1 stats overview
02:15 HumanPSD + TRANSPATH release 2024.1 stats overview
04:48 Short tour to TRANSPATH: pathways, reactions, molecules, complexes
18:18 MATCH Suite for human, mouse, and rat genes analysis. Demo analysis on a Rat dataset of cardio fibroblasts (GSE238154)
23:07 Is there a preference to the type of entities when represented in a pathway? E.g. do you prefer evidence level entities to pathway or semantic level entities
25:44 Importing data from GSE238154 to the geneXplain platform (read counts); identification of DEGs with Limma and filters
29:29 Analysis of upregulated genes from GSE238154 with MATCH Suite. Applying different analysis launch parameters: narrowing down the profile to certain TFs and functional categories
40:43 TRANSPATH database content overview. PDL1 activation pathway.
52:52 Applying Genome Enhancer tool for the analysis of colorectal cancer dataset (PMID: 38520088). Pathway analysis, identification of master regulators, associated drug targets and treatments for potentially overcoming the resistance to PD-L1 therapy.
Don’t do pathway analysis without TRANSFAC
18 June 2024, the twenty seventh “Coffee break with TRANSFAC” session
Click here to view the event details
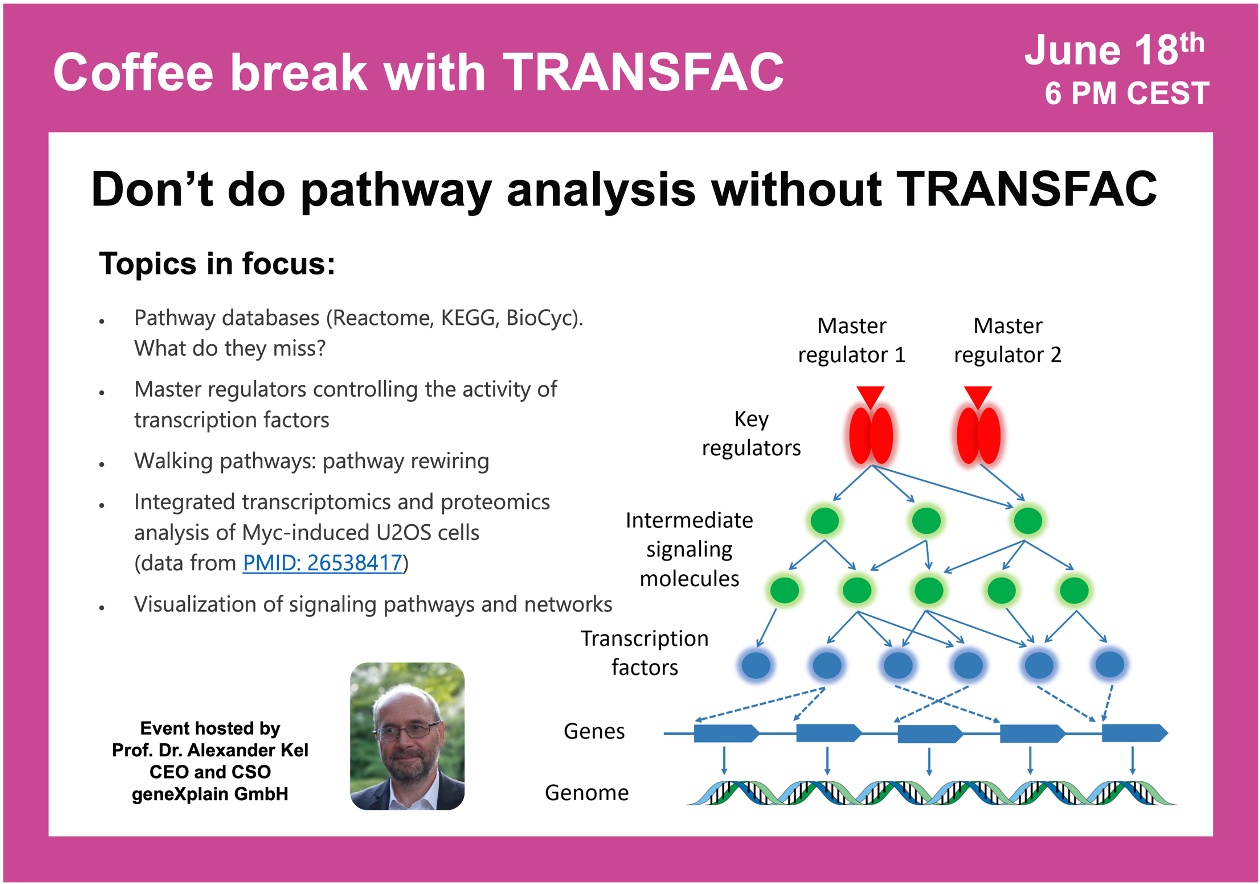
Your questions addressed within this session:
00:56 Why prediction of interactions between TFs and their binding sites needs to be done if you don’t want to miss the molecular mechanisms you are studying during pathway analysis
03:46 Example from GSE222916. Gene expression of fibroblasts treated with TGFβ (transformation to fibrosis)
10:20 Pathway analysis tools on the example of REACTOME, KEGG, PANTHER DB and CPDR from Max Planck Institute for Molecular Genetics – allowing to identify secondary effects (downstream analysis) but not the causative targets of the observed process (upstream analysis)
14:57 Why standard pathway enrichment analysis won’t allow you to identify the original changes? Variability of the target gene sets depending on the tissue or cell type. Necessity of predicting the target genes for the given conditions (pathways stop at the level of TFs)
19:27 Key principles of search for master regulators
29:39 The concept of Walking Pathways (pathway rewiring)
33:17 Genome Enhancer tool for master regulator search (Upstream Analysis)
35:02 Launching Genome Enhancer on raw gene counts data from GSE222916
40:45 Genome Enhancer analysis results overview
42:05 Do we give input to Genome Enhancer as raw counts or the upregulated and downregulated genes separately?
48:51 Another example where two omics types (transcriptomics and proteomics) are integrated in one analysis (based on the data from PMID 26538417). Osteosarcoma was studied at Myc activation. Proteome appeared to be correlating with transcriptome in this experiment.
01:00:08 Is it possible to analyze the structure of promoter, like which sequence is necessary and which is dispensable?
Download TRANSFAC and do whatever you like
28 May 2024, the twenty sixth “Coffee break with TRANSFAC” session
Click here to view the event details

Your questions addressed within this session:
01:56 TRANSFAC flat file download structure and package overview
10:24 Differences between TRANSFAC download and TRANSFAC omics
16:17 How are different species covered in TRANSFAC download?
19:14 TRANSFAC online packages overview
19:46 TRANSFAC download archives structure and database statistics
24:48 What will you see having downloaded and unpacked TRANSFAC download
36:31 Uploading of TRANSFAC JSON files into a relational DB (PostgreSQL)
27:17 Is it possible to filter data based on the experimental condition of interest? E.g. the binding TF, cell type, organism or treatment, in order to retrieve the list of binding sites found in the desired condition(s)?
31:38 Interlinkage of TRANSFAC download entries
38:45 MATCH tool in TRANSFAC download package
40:52 What do you need to have on your computer to be able to run MATCH in TRANSFAC download?
43:56 MATCH library applications
46:57 Conversion from TRANSFAC matrix format to other formats
48:25 TRANSFAC download demo (using the command line)
56:11 Profile creation: creating a collection of matrices for further launching the MATCH search on them
01:02:32 Is the matrix.json file created with the command different from the one present in the json subfolder of the database? (if using the full matrix.dat)
01:04:08 Launching MATCH from command line interface and viewing the site search results
01:09:59 In the TRANSFAC DB, do we have data “catalogued separately” for: a) specific disease-causing genes and their TFs (e.g. diabetes, hypertension or different cancer types); b) for certain viral proteins interacting to human genes (for instance ACE2-SARS-CoV-2 spike protein and the binding TFs to human ACE gene)?
01:13:47 What is missing in TRANSFAC download (check this comparison table)
01:16:26 How fast/productive is MATCH in TRANSFAC download? How much time is needed for the analysis to complete?
01:19:40 Can MATCH be run in parallel?
How to find tissue-specific transcription factor target genes?
21 May 2024, the twenty fifth “Coffee break with TRANSFAC” session
Click here to view the event details
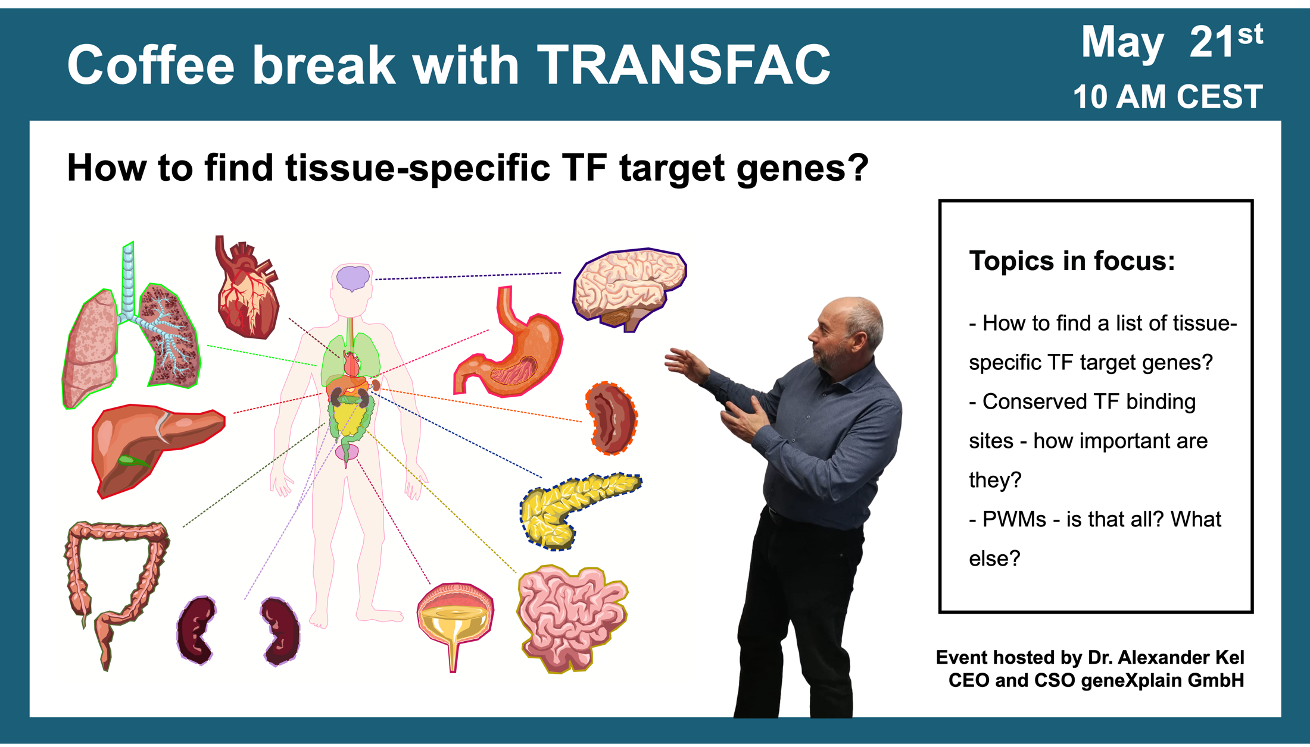
Your questions addressed within this session:
01:42 If you have a list of genes that you are studying and these genes are expressed in a certain tissue, which transcription factors are regulating them?
02:39 Example of artificial tissue construction for heart muscle (GSE 261628) RNA-seq of cardiomyocytes with fibroblasts cells vs. cardiomyocytes with fibroblasts and with macrophages cells
04:55 Finding upregulated genes and analyzing their promoters
06:58 Promoter analysis of genes with a focus on their expression patterns
11:11 Logging in to the geneXplain platform
13:50 Upload of an Excel file with gene counts for identification of DEGs
16:57 Analysis of regulatory regions of selected genes using TRANSFAC (promoter analysis)
23:48 Observing the enriched motifs found in the promoters of the studied genes. Site enrichment and sequence enrichment.
26:37 Finding actual TFs corresponding to the found motifs by applying the gene expression filter (with a focus on heart muscle expression)
30:36 MATCH Suite gene set analysis – interface overview
31:40 – Different types of TFs and similarity of their binding motifs within the same family on the example of IRF factors. Difference in their expression levels in various tissues.
34:34 Constructing a tissue-specific gene set with MATCH Suite
38:42 Gene set analysis results overview in MATCH Suite
45:45 Heatmap visualization of motifs specific for a particular subset of GO terms
49:51 Can analysis of other organisms (not human) be performed using the TRANSFAC database? Analysis of model organisms in the geneXplain platform.
53:06 In the shown tables we saw that several matrices were linked to one factor. How can this be interpreted? Can there be two or more motifs linked to a specific factor?
58:58 Genome Enhancer and TRANSFAC database brief overview
ATAC-seq, CUT&RUN and Enhancers
14 May 2024, the twenty fourth “Coffee break with TRANSFAC” session
Click here to view the event details

Your questions addressed within this session:
00:29 Promoters and enhancers: control of gene expression
07:33 CUT&RUN sequencing
12:41 Peak calling algorithms
16:05 Acute Myeloid Leukemia (AML) study with a focus on BPTF transcription factor (wild type vs. knockout) GSE226670
20:36 Uploading data to the geneXplain platform directly from the SRA archives
22:07 Mapping of reads from paired end sequencing to the genome; BAM files visualization in the genome browser
29:05 Where transcription factor can bind? Alignment of known sites to positional weight matrices (PWMs). Sequence analysis with PWM.
40:22 Identification of enriched TFBS in ChIP-seq peaks
43:19 What do we mean by ‘joined’ peaks as can be seen in the data panel?
47:38 Diffuse large B-cell lymphoma (DLBCL) study with a focus on FOXO1 knockout: ChIP-seq and CUT&RUN, GSE254405, integrated epigenomics + transcriptomics data analysis using Genome Enhancer
57:16 On which basis we will call a peak as open or close? (spoiler: all peaks are showing open chromatin)
1:06:28 In one slide it was mentioned that CUT&RUN seq is cost-effective compared to ChIP-seq, could you please emphasize this point a bit?
geneXplain platform API
30 April 2024, the twenty third “Coffee break with TRANSFAC” session
Click here to view the event details

Your questions addressed within this session:
02:05 What is API and how is it implemented in the geneXplain platform
10:00 geneXplainR package: where to get the documentation and how to use it
15:16 Using RStudio to run scripts in the geneXplain platform API. Installing geneXplainR library; logging in to the platform; project selection; starting the analysis
20:07 Importing data to your project using the geneXplain platform API on the examples of FASTA files and tracks
32:26 While performing the analysis, do we need to download all the files to our local machine? Or can the script extract the .gz files directly (upload) from databases like Ensembl/UCSC or SRA?
36:20 Uploading file with raw gene counts using the geneXplain platform API. Preparing the meta data description files.
41:38 Launching the workflow for DEGs identification using the geneXplain platform API (counts normalization; running Limma). Filtering of the obtained results and eventual DEGs selection.
52:27 Visualization of gene expression data using the ggplot2 R package
56:10 Running Jupyter Notebook scripts in geneXplain platform API; Python scripts
58:21 Java package for geneXplain platform API. Running command line application. Browsing ready JSON files that can be used for your analysis and constructing your own JSON scripts.
01:05:17 Why to use the geneXplain platform API if you are already happy with R, Python and other solutions for conducting your research?
01:10:08 geneXplain platform API functionality summary
1:13:08 Does the documentation provide details on commands to be used on Linux systems?
Promoter analysis of plants
22 April 2024, the twenty second “Coffee break with TRANSFAC” session
Click here to view the event details

Your questions addressed within this session:
01:39 Purine alkaloids in plants
04:14 Coffee species and their caffeine content
18:07 Transcription regulation in somatic embryogenesis of Coffea Canephora; dataset GSE128888
22:40 Analysis of Coffea canephora dataset (GSE128888) using the geneXplain platform tool
25:21Uploading the coffee genome to the geneXplain platform (as a FASTA file plus gene annotation in the format of a GFF file)
30:03 Calculating differentially expressed genes (DEGs) from RNA-seq data on Coffea canephora (GSE128888) using the geneXplain platform with guided Limma
43:01 Processing of the 157 upregulated genes; extraction of their promoters for further analysis; construction of a background set (genes that didn’t change their expression upon induction)
46:03 Search for transcription factor binding sites (TFBS). TFBS enrichment
50:09 Combinatorial analysis: compositions of TFBS discriminating the promoters of upregulated genes from the non-changed genes
52:54 Biological interpretation of the found transcription factors and their impact on the caffeine content in coffee
01:04:19 Genes producing enzymes for caffeine production: close lookup into their expression levels at early stages of plant development
01:08:30 How is the studied coffee species (Coffea canephora) different from C. arabica?
Your first TRANSFAC analysis
16 April 2024, the twenty first “Coffee break with TRANSFAC” session
Click here to view the event details

Your questions addressed within this session:
00:21 Why do you need to analyze anything using TRANSFAC? Why do you need to predict TFBS?
02:57 Starting TRANSFAC analysis from scratch: TRANSFAC database overview
10:00 Observing the geneXplain platform tools
17:47 Uploading a gene list to the geneXplain platform
23:35 Transcription factors, their binding sites and models for their prediction
31:14 Does TRANSFAC DB include data on all the three layers about a specific TF (means adaptor, co-activator)?
33:08 Motif scanning: how scores are calculated (Sum of the contribution of each nucleotide calculated independently (like in HOMER) versus sum of the contribution of each nucleotide multiplied by an information vector (like in MATCH))
38:20 Calculation of optimized cut-offs for PWMs
43:02 Why a NO set (control) is needed for searching for TFBS. Enrichment analysis.
45:37 Site search on gene set in the geneXplain platform (MATCH): analysis launch and results overview
01:02:27 Visualization of found sites in the genome browser
01:04:10 HOMER and other Galaxy tools in the geneXplain platform
01:07:34 Whatever analysis is done, is there a summary report generated?
01:10:16 Will there be any sessions on miRNA target prediction in future?
Exploring the FREE geneXplain platform account
8 April 2024, the twentieth “Coffee break with TRANSFAC” session
Click here to view the event details

Your questions addressed within this session:
01:12 What is geneXplain platform and how to get a free account in it
02:45 geneXplain platform start page and tree area overview (databases, projects, methods, and workflows)
09:25 Workflow overview and workflow editing
11:58 Differentially expressed genes (DEGs) identification in the geneXplain platform interface
18:34 Exploring various methods on the example of a list of DEGs: convert table (from Ensembl gene IDs to UniProt IDs); filter table (by LogFC)
21:41 BED file upload and visualization on track. Opening of the peaks track in the table view. Converting from hg19 to hg38. ChIP-seq: functional classification of target genes.
29:17 How do I upload bulk RNA-seq files for analysis?
31:42 Is there a history stored for all the analysis conditions that are used?
33:17 No-coding bioinformatics academy from geneXplain
34:41 Sharing your projects with other people
36:05 Site search analysis in the geneXplain platform free account (including GTRD, HOCOMOCO, HOMER)
45:57 Combinatorial analysis of TFBS: MEALR and CMA algorithms
49:32 Pathway analysis and network analysis in the geneXplain platform free account (REACTOME, HumanCyc, GeneWays). Regulator search (master regulators).
56:00 Statistical analysis (Limma, PCA, Heatmaps, t-SNE)
58:56 Do you have any tools for combining technical / biological replicates in a single data set to work with?
01:04:35 geneXplain platform API
Your income and your DNA
2 April 2024, the nineteenth “Coffee break with TRANSFAC” session
Click here to view the event details

Your questions addressed within this session:
00:00 Is your level of income written in your DNA?
00:23 Dataset from Nature paper showing 120 income associated SNPs and their significance. (Hill, W.D., Davies, N.M., Ritchie, S.J. et al. Genome-wide analysis identifies molecular systems and 149 genetic loci associated with income. Nat Commun 10, 5741 (2019). https://www.nature.com/articles/s41467-019-13585-5)
08:40 Upload of the income associated SNPs to Genome Enhancer
13:20 Site gains and site losses as a consequence of SNPs
19:25 Search for TFBS with TRANSFAC. Effects of SNPs on TFBS. Score differences.
21:31 Motif enrichment analysis in the areas around SNPs
23:40 Selection of genes, expression of which is regulated by the studied SNPs
26:46 Converting the SNPs table to positions in the genome. Conversion of SNPs from hg19 build to hg38 build using the Liftover method inside the geneXplain platform
30:30 Converting the list of SNPs into a gene list (selection of ‘nearby’ genes, but how about distant enhancers?)
34:44 Functional classification of selected genes by expression values in different tissues
39:52 Eventual gene list: which SNPs can influence the expression of nearby genes that are known to be expressed in the brain
40:25 Genome Enhancer: analysis launch
43:00 Genome Enhancer: exploring the analysis report
44:48 How are the SNPs affecting the TFBS; site gain and site loss p-values
46:48 Interpretation of master regulators diagram (master-regulators of SNPs that correlate with income)
49:25 Is trust also regulated by oxytocin?
53:33 GWAS generally tells about “collaborating” SNPs resulting in a particular trait. Can TFs form kind of “basis” of such an SNP/trait interplay? Can geneXplain predict these TF particularities?
58:00 Are only SNPs occurring in the over-expressed genes taken into consideration or the % of SNPs in the under-expressed genes also hold importance when we look at the SNPs and genes? (like the brain tissue in the case discussed here)
01:01:56 SNPs found to be correlated with educational attainment in another study (https://youtu.be/crGr3RWuYuM and https://youtu.be/vmx1jjcMTdU) appeared to be distributed among the genome, but in case of income-associated SNPs they appeared to be clustered in one area and connected to a much lower number of genes
MATCH Suite software demo
19 March 2024, the eighteenth “Coffee break with TRANSFAC” session
Click here to view the event details

Your questions addressed within this session:
01:27 geneXplain portal interface brief overview
02:25 Search interface of the TRANSFAC database; locus report, site report, matrix report overview
10:33 Launching MATCH Suite single gene analysis from the TRANSFAC database search interface
16:56 Single gene analysis results overview in MATCH Suite
18:08 MATCH Suite single gene analysis report summary overview
21:16 MATCH Suite single gene analysis pipeline overview (methods used)
31:03 MATCH Suite single gene analysis report details
37:20 MATCH Suite single gene analysis interactive results visualization
43:19 How different MATCH Suite single gene analysis results are, when different analysis conditions are specified for the analysis of the same gene?
57:24 Launching MATCH Suite single gene analysis on various inputs suggested by the audience
01:09:03 Gene set analysis launch from the MATCH Suite interface
01:14:53 MATCH Suite gene set analysis report summary overview
01:16:34 MATCH Suite gene set analysis pipeline overview (methods used)
01:20:54 MATCH Suite gene set analysis report details
01:27:47 MATCH Suite gene set analysis interactive results visualization
01:38:20 How different MATCH Suite gene set analysis results are, when different analysis conditions are specified for the same input gene list?
01:39:55 Launching MATCH Suite gene set analysis from the TRANSFAC database ontology search results
01:47:51 Starting the gene set analysis in MATCH Suite from a gene set constructed on the fly based on the gene expression values in a selected tissue
01:52:07 geneXplain platform introduction and its possible integration with the usage of MATCH Suite
Promoter analysis of model organisms (part 2)
12 March 2024, the seventeenth “Coffee break with TRANSFAC” session
Click here to view the event details
Your questions addressed within this session:
01:11 Number of different positional weight matrices (PWMs) or motifs contained in TRANSFAC for various organisms
02:56 Matrix construction for TRANSFAC database
04:08 Site enrichment analysis using TRANSFAC PWMs
06:07 Composite elements of transcription factors in gene regulation
07:16 Genetic algorithm for identification of TFBS combinations
10:45 geneXplain platform overview
13:03 Example: RNA-seq and ChIP-seq data analysis from Drosophila melanogaster (GEO: GSE149116) Spoiler: human ZKSCAN3 and Drosophila M1BP are functionally homologous transcription factors acting as master transcription factors autophagy regulation! It appeared that transgenically expressed human ZKSCAN3 gene in drosophila started to regulate many drosophila genes binding to the same sites as endogenous M1BP factor.
18:20 Starting an integrated ChIP-seq and RNA-seq analysis inside the geneXplain platform: using DEGs from transgenic drosophila when compared to the wild type, and ChIP-seq peaks for the antibody for human ZKSCAN3 transcription factor from GEO: GSE149116
24:17 Is there a “proper” promoter length for performing the analysis?
28:35 What is the advantage of using the geneXplain platform to see the results of this type of experiment (integrated ChIP-seq and RNA-seq analysis) instead of using the IGV like apps?
32:39 Analysis of DEGs regulation in the geneXplain platform: filtering of a table with DEGs and selecting the ChIP-seq peaks located near the significantly differentially expressed genes using the filter one track by another function
40:38 Finding enriched motifs in tracks constructed as ChIP-seq peaks located around the upregulated genes
45:47 Results overview and interpretation for the identification of enriched motifs in tracks (site search summary and MATCH track)
50:35 Launching the CMA (Composite Module Analyst) analysis, and CMA results overview
57:53 What can TRANSFAC do in relation to enhancer sequences?
01:03:36 Are there also curated СhIP-seq datasets in TRANSFAC that can be used / compared along with own data?
01:07:33 Experimentally proven TFBS in TRANSFAC
01:09:24 The genome used for human is hg38, but is it the canonical version or the whole assembly?
Promoter analysis of model organisms (part 1)
4 March 2024, the sixteenth “Coffee break with TRANSFAC” session
Your questions addressed within this session:
02:37 Model organisms in TRANSFAC database
06:37 geneXplain platform overview: model organisms available in the system by default
09:20 viewing TRANSFAC motifs in the geneXplain platform interface; what is a profile and how to select it
12:08 Construction of a PWM (positional weight matrix). Cutoff and core cutoff.
17:13 Mini pig model for atherosclerosis study
19:03 Importing data to the geneXplain platform via ftp
21:35 Principles of site enrichment analysis using the TRANSFAC database
23:59 Upload of a new genome to the geneXplain platform (from NCBI)
27:33 Genome browser visualization of the newly uploaded genome
32:47 Identification of DEGs (differentially expressed genes) with Limma; filtering of the obtained results
39:34 Extraction of promoters of the pig genes using the “process track with sites” function of the geneXplain platform and the coordinates of pig genes taken from the MART database (EBI)
43:23 Search for enriched TFBS using the Site search on gene set function of the geneXplain platform
49:40 Finding combinations of motifs belonging to TFs working together using the CMA (Composite Module Analyst)
55:05 Upstream analysis with feedback loops – results overview
01:02:26 Can you start the analysis from differentially expressed genes (Limma output)?
01:07:42 Converting between different accession numbers in the geneXplain platform
01:11:02 Overview of the integrated HumanPSD+TRANSPATH+TRANSFAC database interface
6 February 2024, the fifteenth “Coffee break with TRANSFAC” session
How to find tissue-specific TF target genes Click here to view the event details

Your questions addressed within this session:
0:44 How to find all target genes of NFkB transcription factor? Clue: in which tissue?
02:17 MEF-2A transcription factor and its expression profile
04:20 Which TFs are regulating my genes specifically expressed in muscle tissue
05:42 MATCH Suite gene set analysis launch
09:41 MATCH Suite gene set analysis results overview
22:57 MATCH Suite gene set analysis report overview
26:31 Tissue-specific gene regulation
28:30 TFClass transcription factors classification
30:50 Expression patterns of transcription factors coming from one family
33:55 Constructing tissue specific gene set in MATCH Suite
36:26 Gene list optimization by GO terms in MATCH Suite
41:39 Heatmap of GO terms and TF motifs produced by MATCH Suite
44:55 MATCH Suite results overview from the geneXplain platform interface
48:41 In my research area (plant pathogenesis) TFs are not enough available in Jaspar. Can I proceed with such species when experimentally validated TFs are not available in the database?
52:31 We found an unknown Zinc finger protein. Can I use your tool to predict the target genes?
55:20 Any cross-reference data base on transcription factors?
57:29 If we are analyzing a TF gene and the question is to find the downstream genes of this TF in genome of that organism. How the downstream genes can be identified? (check out this other video demonstrating how all TFBS of one transcription factor can be found)
23 January 2024, the fourteenth “Coffee break with TRANSFAC” session
Your first TRANSFAC analysis Click here to view the event details

Your questions addressed within this session:
02:32 Gene list analysis in the geneXplain platform (search for TFBS in promoters of genes in focus)
04:48 Upload of table with differentially expressed genes (DEGs) to the geneXplain platform
07:55 Saving a single gene from a gene list for further single gene promoter analysis
08:33 Prediction of transcription factor binding sites (TFBS) in the promoter of a single gene (site search on gene set with 1 gene in the list)
14:30 Visualization of found sites in the promoter model of the studied gene
16:25 Opening the track of found sites in the full genome browser visualization
17:37 Dragging and dropping genes, repeats, and variations tracks to the genome browser visualization
19:19 Export of the obtained results: saving the table of found sites and export of found sites as a BED file
22:29 – What do you mean by property score and core score? Does it give p-values of the predicted transcription factors? On what basis can we select the TFs?
23:41 The model of eucaryotic gene regulation
28:37 How are the matrices built? Additive model of sites prediction
30:41 From count matrix to the frequency (probability) matrix. Log-odds score of sites (used in HOMER)
34:08 MATCH site score: multiplication by the information vector
36:01 The score cutoffs selection (which cutoffs to select for your MATCH analysis)
37:44 The profile used in MATCH analysis: collection of matrices and their cutoffs
40:38 Comparison of MATCH algorithm with log-odds or additive algorithm
41:31 Protein-protein interactions compensating the actual binding energy of individual site. Why cutoffs should not be fixed at all and can be automatically found by the algorithm
44:38 Working with gene sets describing the studied condition, e.g. DEGs and non-changed genes that didn’t respond to the condition (yes and no sequences)
46:06 Automatic cutoffs identification that maximize the difference between the sites found in yes and no sets
47:15 Site search on gene set using a No set (non-changed genes)
47:55 Profiles used for site search
51:51 Is it possible to search for motifs for several TFs taking into account their interaction?
53:17 Overview of the output table of gene set analysis with No set and cutoffs optimization
54:53 Visualization of found sites in the models of promoters of the genes from the input gene set
59:26 Is the promoter browsing interval fixed? (1000 bp upstream and 100 bp downstream the TSS?)
01:04:18 Is it possible to find TFs if we don’t have any ChIP-seq data for the particular gene?
01:07:46 How do you backtrack a particular TF from a matrix predicted with your tools? How do I know which protein is actually regulating my gene? What are the filters I can apply to all TFs corresponding to that matrix in order to select the most probable one for my case?
01:12:56 On the basis of only the score, how can you select the TFs, as TRANSFAC gives a huge list of TFs, how to filter it?

Your questions addressed within this session:
00:56 ATAC-seq data: what is it and how is it processed?
05:05 The model of intracellular gene regulation; master regulators
07:17 Algorithms for ATAC-seq data analysis
08:05 Desmoplastic small-round-cell tumor: GSE226670 dataset (ATAC-seq + RNA-seq)
10:55 SRA, table data, and BED files upload to the Genome Enhancer
18:19 Annotation diagram construction in Genome Enhancer
20:48 Analysis launch in Genome Enhancer: disease selection and specification of conditions for comparison
22:47 Genome Enhancer results: report overview
29:50 Transcription factor binding sites in ATAC-seq peaks
36:45 Search for master regulators; positive and negative feedback loops
42:21 Selection of prospective drug targets and associated treatments
44:30 Genome Enhancer results visualization in the geneXplain platform interface
52:41 How can quantitative information be applied in ATAC-seq peaks analysis? Interpretation of the ATAC-seq peaks height: how often a particular location is accessible for the enzyme? Selection of motifs appearing more frequently in high peaks and less frequently in low peaks. Alternative quantitative information: expression level of a gene.

Your questions addressed within this session:
00:37 ATAC-seq data – what is it?
04:18 How ATAC-seq data is analyzed
06:50 The general approach towards upstream analysis
11:23 Algorithms for ATAC-seq data analysis, peak calling approaches
13:53 GSE226670 dataset on acute myeloid leukemia cell line: integrated RNA-seq and ATAC-seq analysis devoted to BPTF knock-out compared to wild type
18:13 Analysis of GSE226670 ATAC-seq and RNA-seq data using the geneXplain platform
19:17 SRA data upload to the geneXplain platform using the SRA ID
21:21 FASTQ files mapping to the genome in the geneXplain platform (with Bowtie2)
22:59 BAM file visualization on genome browser in the geneXplain platform
24:24 Peak calling with MACS2 from a BAM file in the geneXplain platform
27:43 Search for transcription factor binding sites (TFBS); search for enriched TFBS in ATAC-seq peaks areas
29:42 Uniting all peaks from one experiment (join tracks function of the geneXplain platform)
31:33 Finding peaks from the wild type cells that are not present in the knock-out experiment (intersect tracks function of the geneXplain platform)
32:41 Epigenomics data analysis in the geneXplain platform: site search with TRANSFAC
36:06 Site search results visualization and interpretation
44:00 Genome Enhancer application for integrated ATAC-seq and RNA-seq data analysis. Analysis launch
48:56 Genome Enhancer analysis report. Enriched transcription factors and key master regulators. Intersection of peaks from the wild type cells with the upregulated genes
54:57 The YY1 (Yin Yang) binding sites balance; the Yin Yang factor with controversial regulatory functions
58:40 Further steps after TFBS analysis: master regulators in Genome Enhancer report and prospective drugs overview

Your questions addressed within this session:
02:53 The basic model of gene functioning and regulation
05:24 Brief overview of transcription factor binding sits (TFBS) prediction
09:06 How to find master regulators that control the activity of identified transcription factors
14:18 Walking pathways: pathway rewiring
19:27 GeneXplain platform brief interface overview
20:52 Example of gene expression analysis data from GSE66789 (normalized counts – FPKM – from osteosarcoma cell lines with induced Myc gene) + proteomics data (logFC of protein expression in Myc induced cells compared to controls)
23:25 Data upload to the geneXplain platform
25:47 Finding differentially expressed genes (DEGs) in the geneXplain platform interface
27:11 Filtering of DEGs, selection of upregulated genes
28:31 For RNA-seq data analysis can TPM be used as well? (instead of FPKM)
29:26 Construction of the control set of genes (needed for search of enriched motifs in promoters (TFBS search)
31:11 Finding binding motifs
35:21 Overview of the site search tabular results; Yes-No ratio
38:18 Genome Browser visualization of the found motifs
43:35 Upstream analysis: searching upstream of the identified transcription factors. Identification of master regulators.
53:49 The constructed network is generated by the means of the literature search or is it from the specific experiment?
55:35 Visualization of TRANSPATH pathways in Cytoscape
59:20 Different weights of the network proteins based on the betweenness centrality

Your questions addressed within this session:
01:57 What is the way from transcription factors to pathways? How do transcription factors operate?
03:15 What happens after we have identified transcription factor binding sites (TFBS)?
06:33 Prediction of TFBS in promoters of genes using positional weight matrices (PWMs)
08:20 Combinations of transcription factors, Composite Module Analyst (CMA)
10:48 Search for master regulators
21:15 Walking pathways
31:22 Loading of RNA-seq data (normalized counts) to the geneXplain platform
34:50 Filtering of genes; selection of up and down regulated genes and non-changed genes
36:11 Identification of enriched motifs in promoters of genes using TRANSFAC
41:06 What are the transcription factors regulating your genes (after finding the enriched motifs)
44:33 Going upstream from the identified transcription factors
45:01 Regulator search: how to find master regulators
50:10 Network visualization of master regulators
51:03 Annotating the visualization diagram with expression data
51:54 Applying different layouts to the constructed network
52:55 Opening the TRANSPATH network in Cytoscape
55:45 Can own interaction data be introduced by the user? REACTOME, Recon, GeneWays
58:20 Can you mix different databases? Adding a file with different interactions from various databases
01:01:32 Is it possible to construct the master regulator-TFs network without proteomics context? Are there any reconstruction examples of this type network for microorganisms?
01:03:53 How can we plot the signaling pathway for tuberculosis host hub genes (or any genes) using the geneXplain platform?
01:09:06 Is it possible to discover new regulatory pathways based on expression data?
Your questions addressed within this session:
00:20 Gene regulatory networks – what are they and how to construct them
21:12 MATCH Suite and gene regulatory networks based on predictions of transcription factor binding sites
27:35 MATCH Suite results visualization: constructing gene regulatory networks using Python* in Jupiter notebook (geneXplain platform API)
*The Python code shown in the online demo is available here
40:42 From which online software I can get the sequences of transcription factors?
50:30 Can TRANSFAC be applied for other species, e.g. drosophila or plants? What are the limitations?

Your questions addressed within this session:
00:02 Introduction: TRANSFAC in Cancer research
00:56 World Brain Tumor day
01:45 Transcription factors: how do they regulate their target genes
02:21 Positional weight matrices – TFBS model
03:07 Searching for TFBS enrichment
03:48 Composite complexes of transcription factors
04:47 Search for Master Regulators
06:26 Intracellular signal transduction: complex cascades consisting of signaling reactions
08:18 Construction of potential network of possible reactions within the cell
09:01 Modeling of regulation of a particular set of genes
11:59 Algorithm of Master Regulators searc
13:42 Integration of context protein expression information in the search for Master Regulators
15:23 Verification of Master Regulators quality
18:08 Walking pathways concept
20:35 How to select the “true” Master Regulator
22:08 Therapeutic targets and biomarkers of the studied processes
22:59 Methylation marks located in regulatory regions of Master Regulator genes are good diagnostic or prognostic biomarkers
23:18 The Upstream Analysis concept (integrated promoter and pathway analysis)
23:58 Genome Enhancer – the fully automated pipeline for prospective drug target identification
26:43 Analysis of Glioblastoma short-term survival patients vs. long-term survival patients using Genome Enhancer (this analysis resulted in the following publication: IGFBP2 Is a Potential Master Regulator Driving the Dysregulated Gene Network Responsible for Short Survival in Glioblastoma Multiforme)
41:47 Switch from Genome Enhancer to the geneXplain platform interface with extended functionality. Mapping of prospective Master Regulators to diseases in which they are known biomarkers.
47:38 Immunotherapy sensitivity prediction for Glioblastoma patients
51:26 Are there transcription factor and enhancer databases for nonhuman primates?
56:30 Is there a classical publication involving geneXplain related to the topic?
Yes, please check those on Glioblastoma: (1) and (2), this one on the Walking Pathways concept, and other publications that can be found here.
58:57 Can we check these results with the data on mutations in samples? Won’t this help find up- or down-regulated genes with respect to their mutation profile?
01:04:53 If I have RNA-seq data for my patient, which additional omics data could improve the analysis with your tool effectively? E.g. variants, protein expression, methylation?

Your questions addressed within this session:
02:47 TRANSFAC brief overview
05:08 Site enrichment analysis
07:59 Site combinations: complexes of transcription factor binding sites; composite modules
14:27 TRANSFAC application for different model organisms
21:27 Example on drosophila genes analysis from GSE149116 – combination of RNA-seq and ChIP-seq data
22:31 What are the target genes of my transcription factor? Is such question correct?
23:16 Human TF was put as a construct and expressed in Drosophila –> Human TF started regulating the drosophila genes! Wow!
25:29 Different experiment types being united in one analysis: intersection of RNA-seq data with ChIP-seq data
28:25 Drosophila data analysis in the geneXplain platform (on the example of GSE149116 dataset)
41:02 How site search summary looks like in the geneXplain platform
42:21 Identification of “master” transcription factors working together in combinations; CMA (Composite Module Analyst) analysis in the geneXplain platform
46:44 Can we use other genomes (other than human) for TFBS analysis with TRANSFAC in the geneXplain platform
47:15 How to upload the palm genome to the geneXplain platform
50:33 Extraction of promoters of genes responsible for palm resistance to different bacteria or infections. “Master” transcription factors regulating the resistant genes in the palm.
54:35 What are the “standard” genomes in the geneXplain platform
56:01 How can I find master transcription factors that regulate specific pathways in Arabidopsis
58:17 Which profile (collection of PWMs – positional weight matrices) to use for the analysis?
Your questions addressed within this session:
- Mutations in cancer: how to analyze them using TRANSFAC?
- Introduction to Genome Enhancer – an automatized pipeline for multi-omics data analysis. Analysis of cancer mutations using Genome Enhancer.
- Lung cancer cell lines analysis using Genome Enhancer. A more detailed video devoted to this example can be found here.
- By which authority is Genome Enhancer certified for application to patient data in hospital?
- Mutations located in non-coding regions of genes: can they destroy or create TFBS?
- Can we predict the effect of these gene mutations on drug binding to targets?
- Analysis of binding sites compositions: search for co-factors – transcription factor binding sites (TFBS) working together. Identification of TFBS compositions around clusters of mutations when compared to regions without mutations.
- Switch to the geneXplain platform perspective: a more detailed view on results produced by Genome Enhancer pipeline in regards to the TFBS analysis.
- Brief overview of the next sections of Genome Enhancer report: pathway analysis, identification of master-regulators in networks and selection of prospective drug targets and associated treatments.
Your questions addressed within this session:
- SNP data analysis: motifs destroyed and created by SNPs – how to find them and what is their effect?
- Overview of the geneXplain platform interface
- Analyzing SNP data with the geneXplain platform (Variant Analysis)
- Is it possible to use TRANSFAC to calculate p-values for binding with a specific allele at the SNP and to rank predicted TFs based on their binding score?

Your questions addressed within this session:
- De novo motifs. What are they? How to find and use them? (short intro)
- Why are there so many motifs (PWMs – positional weight matrices) collected for 1 transcription factor? All of them are different. Which one is “true”? (spoiler: they ALL are!)
- Searching for de novo motifs: when and why do we need new motifs? How are they discovered?
- ChIPMunk motif discovery tool – what is it?
- De novo motif search using the geneXplain platform
- Can I perform TFBS search with TRANSFAC alone?
- Can geneXplain platform find mutation in the intron (when compared with reference sequence, e.g. hg38)?

Your questions addressed within this session:
- How to compute transcription factor binding affinity?
- What is the difference between the 5’ prime and the 3’ prime promoters?
- I have two genes with overlapping promoters, but different TFBS, can you comment on this?
- If I design a minimal promoter, what would be the basis for screening and selecting the best TFBS to include in my minimal promoter if I use the MATCH Suite tool? In brief, how to prioritize these TFs to select the most relevant of them?
- Could you explain what is the difference between p-values in the MATCH Suite analysis report?
- For one TF there can be numerous PWMs defined by a number in the name such as V$AP1_02. Does this number correspond to a new version of the PWM based on the alignment of known TFBS? Do you advise to use the last PWM built on the alignment of known binding sites as the most updated one or do we need to use all PWMs for a TF to perform the prediction for a particular TF?
- Alexander, as I understand, the affinity score(s) is a floating number. How does the program define the cut-off (maybe I missed that) – every time depending on the experiment, or it is set up on an average basis?
- Is it important to have cell specific analysis and not only tissue or organ specific one?

Your questions addressed within this session:
- TFBS prediction in DNA based on positional weight matrices
- How to define the cutoffs for binding sites scores
- In calculating FP and FN you need to know the true binding sites. How are they determined?
- I have some questions about my data derived from the TRANSFAC 2012 professional matrices. 1) Some of the genes of interest have binding sites for IRF (without further specification of the subtype), STAT (again, without further specification of the subtype). Can you explain what this means? 2) Some other genes have binding sites for STAT1STAT1 – does this mean STAT1 dimer, also known as the GAS element?
- I am interested to find out what TFs, promoter & enhancer regions, and epigenetic signatures are relevant for the regulation of specific genes in rat hippocampal pyramidal and inhibitory neurons. I know this is not an easy task, and needs not only a bioinformatic but also an experimental approach. I am not aware how transcriptome (RNAseq) and epigenetic (ATAC-seq) data are integrated in the TRANSFAC platform and whether TRANSFAC could help in refining this.

Your questions addressed within this session:
- How can I start a TRANSFAC bioinformatics analysis? What types of data do I need?
- What is the significance of a transcription factor binding site on plus or minus strand of the gene?
- New transcription factors – do you survey those all the time? How many targets are necessary for inclusion of one (creation of a matrix) in the program?
- What advantages does TRANSFAC have over the HOCOMOCO and JASPAR databases?
- Regarding TF binding analysis: even after we get a putative motif for TFs, how many TF could possibly bind at a single site on the genome, depending on a significance cut-off, I find TF binding sites from 2-3 to almost 40-50 at the same location. How do we filter the noise or get accurate results?
- Is it possible to use directly gene sequences to search TFBS? For example sequences from Ensembl database? FASTA file?
- Either on + strand, or on – strand, one should read the binding site from 5′ end to 3′ end, is it correct?
- Can a gene have two active promoters and corresponding TSS in the same cell?
- In order to investigate how much a genetic mutation could affect a TF binding site, is it possible that such a mutation could affect multiple TFs binding at that region? Would it be advisable to focus only on TFs based on their binding score?
- The co-occurrence of TFs proposed by TRANSFAC is based on the knowledge coming from different experiments, so what are the chances of co-occurrence actually?